Caso Clínico:
HISTORIA CLINICA
Antecedentes Personales: Exfumador.
Actividad laboral: Trabajó en la banca.
Antecedentes familiares: Sin interés. No antecedentes de enfermedades neurodegenerativas.
Varón de 60 años, que presenta un cuadro lentamente progresivo de 3 años de evolución de dificultad para caminar, lentitud en el habla y cambios de comportamiento (irritable, adquirió nuevas "manías" como gastar dinero desproporcionadamente, atracones de comida...) y de memoria (dificultad para atender sus finanzas, para manejar las tarjetas).
En el último año el cuadro empeoró notablemente afectando a la continencia de esfínteres, a las actividades cotidianas (necesita ayuda para todo salvo comer) y a la marcha, en un mes pasó de caminar con bastón, aunque con postura anormal del tronco y las extremidades izquierdas a no poder mantenerse de pie ni caminar por grave inestabilidad, aun con un punto de apoyo.
EXPLORACION CLINICA
E. Neurológica:
Amimia facial, disminución de parpadeo, lentitud en el inicio de sacadas. Bradicinesia grave generalizada de predominio izquierdo. Grasping bilateral y reflejos de liberación frontal.
Rigidez paratónica. Temblor postural irregular de predominio izquierdo. Cuando podía caminar, presentaba una hemidistonía izquierda con abducción y extensión de EE en la marcha. En el momento actual, abolición de reflejos posturales y apraxia de la marcha e imantación que impiden la bipedestación estable.
E. Neuropsicológica:
Inhibido, negativista, oposicionista. Lenguaje escaso, agramático, hipofónico, bradilalia, déficit de comprensión de órdenes complejas, desorientación temporoespacial, buena atención en series inversas, mala fijación (1/3). Nominación, lectura y repetición conservadas. Dispraxia gestual e ideomotora prominente de predominio en ESI.
PRUEBAS COMPLEMENTARIAS
- Analítica: sin alteraciones relevantes,
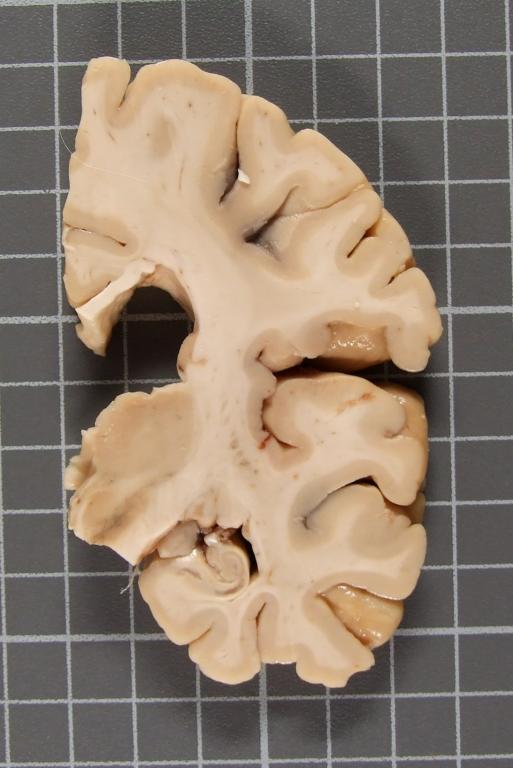
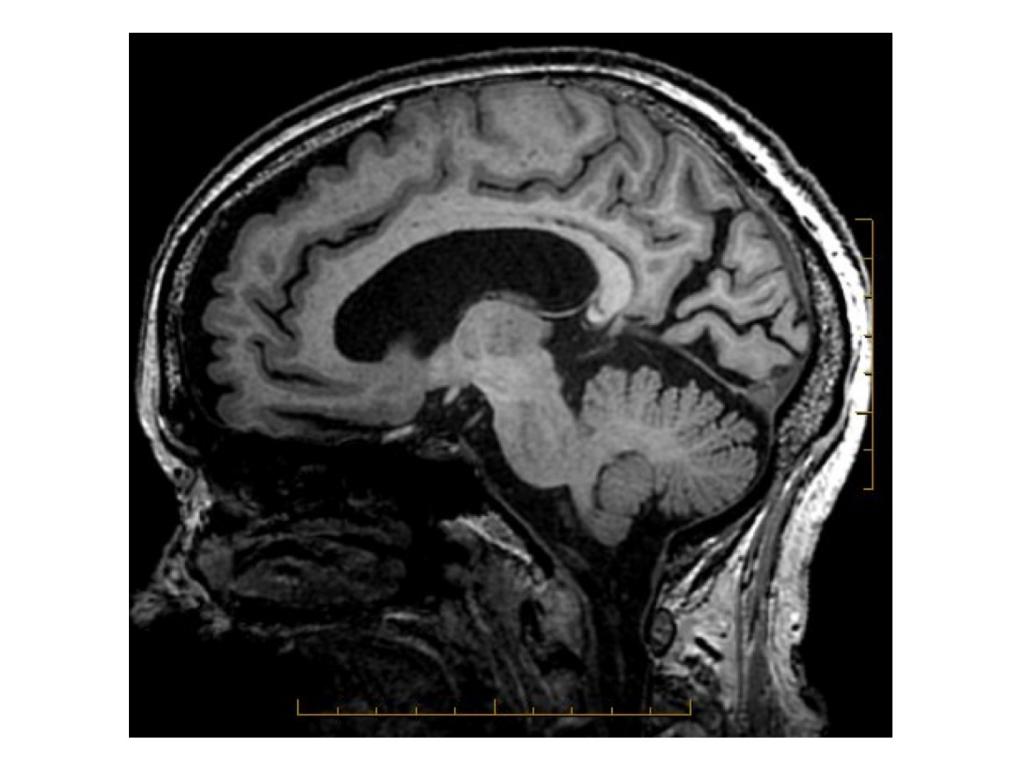
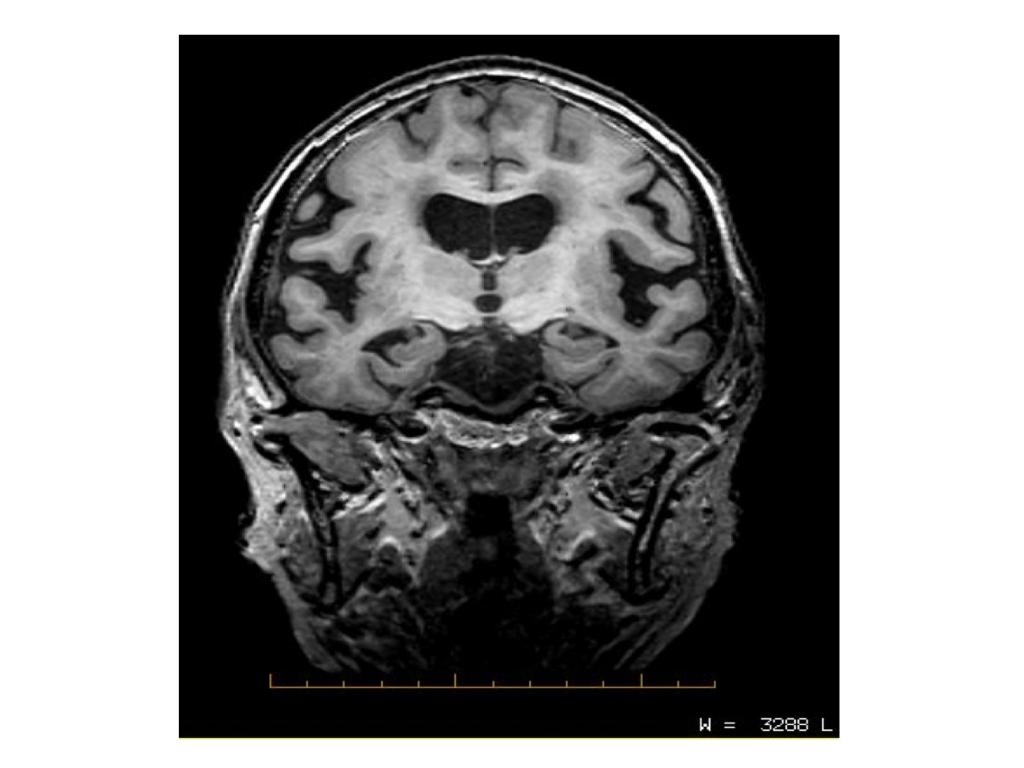
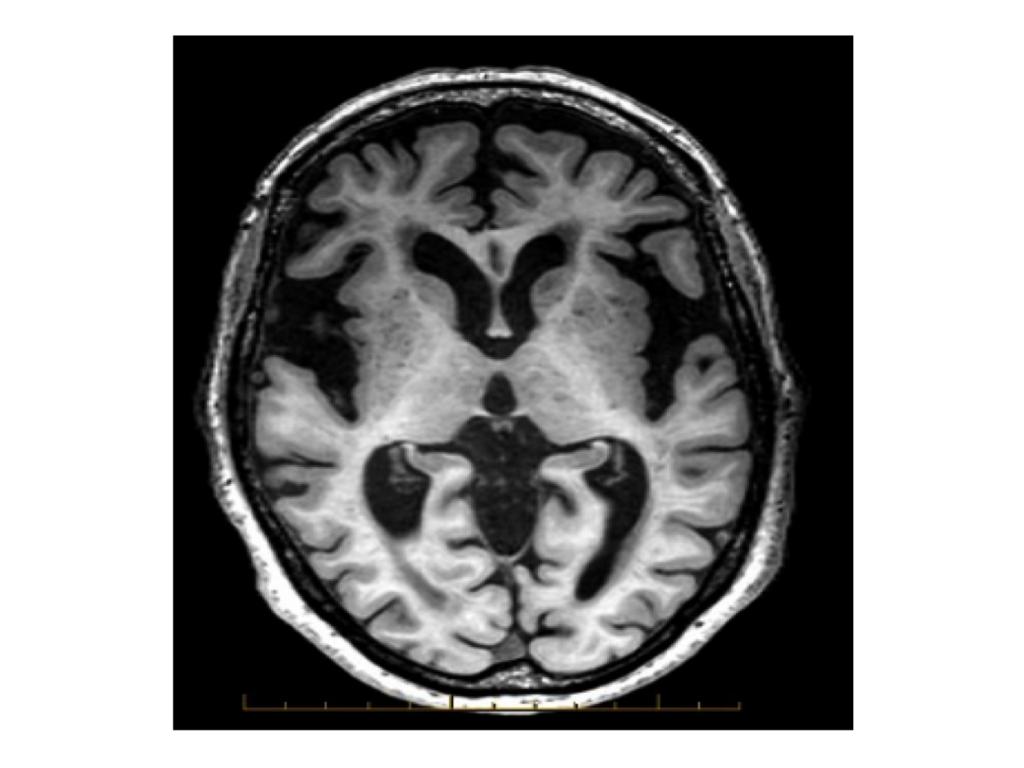
- RM craneal: informada como atrofia córtico subcortical inespecífica.
- DATSCAN-I123 e IBZM-I123:
Hipocaptación en ambos putamenes, leve en el izquierdo y más moderada en el derecho. Fijación conservada en caudados. Compatible con patologia de sistema dopaminergico presinaptico.
Estudio cerebral realizado con trazador de receptores dopaminérgicos D2 marcado con I123 y técnica de SPECT que muestra: Fijación moderada en ambos estriados, con captación extraganglionar inespecífica. Compatible con sistema dopaminergico postsinaptico indemne parcialmente bloqueado por tratamiento agonista.
CONCLUSIÓN: COMPATIBLE CON ENF. DE PARKINSON (VERSUS PARKINSONISMO)
EVOLUCION:
A tenor de la historia y las pruebas complementarias realizadas se inició tratamiento con un agonista dopaminérgico y levodopa, sin beneficio (salvo en el habla, que ciertamente mejoró), ni fluctuaciones en la evolución clínica del cuadro.
El insomnio grave con agitación nocturna, con beneficio parcial con clonazepam.
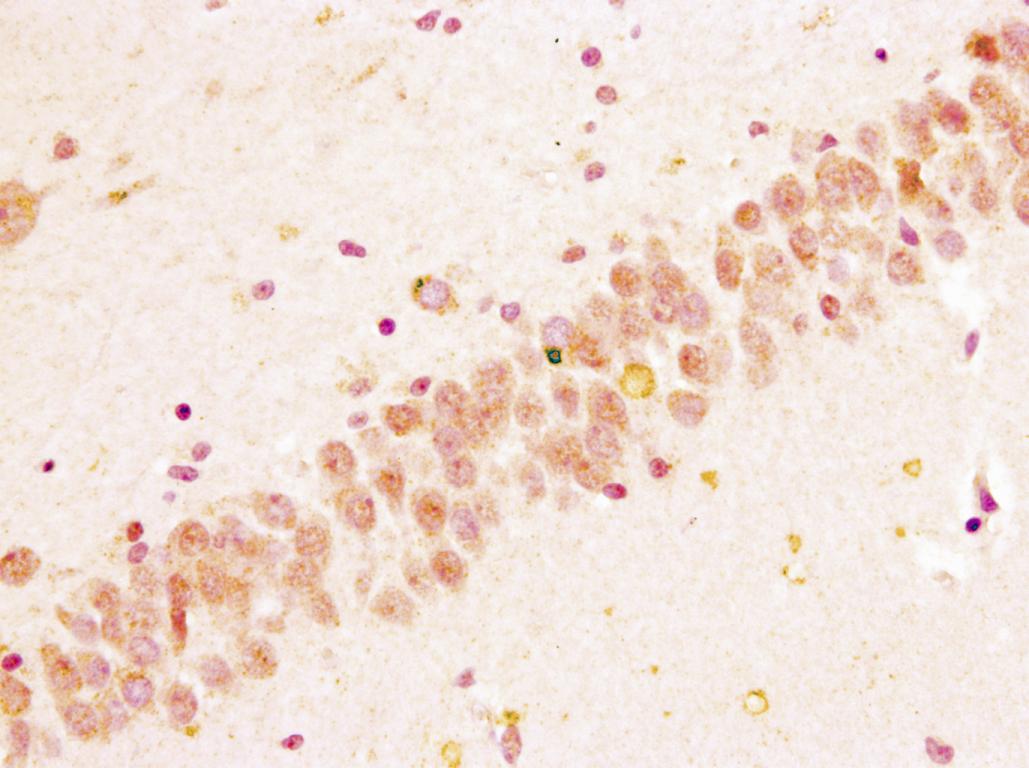
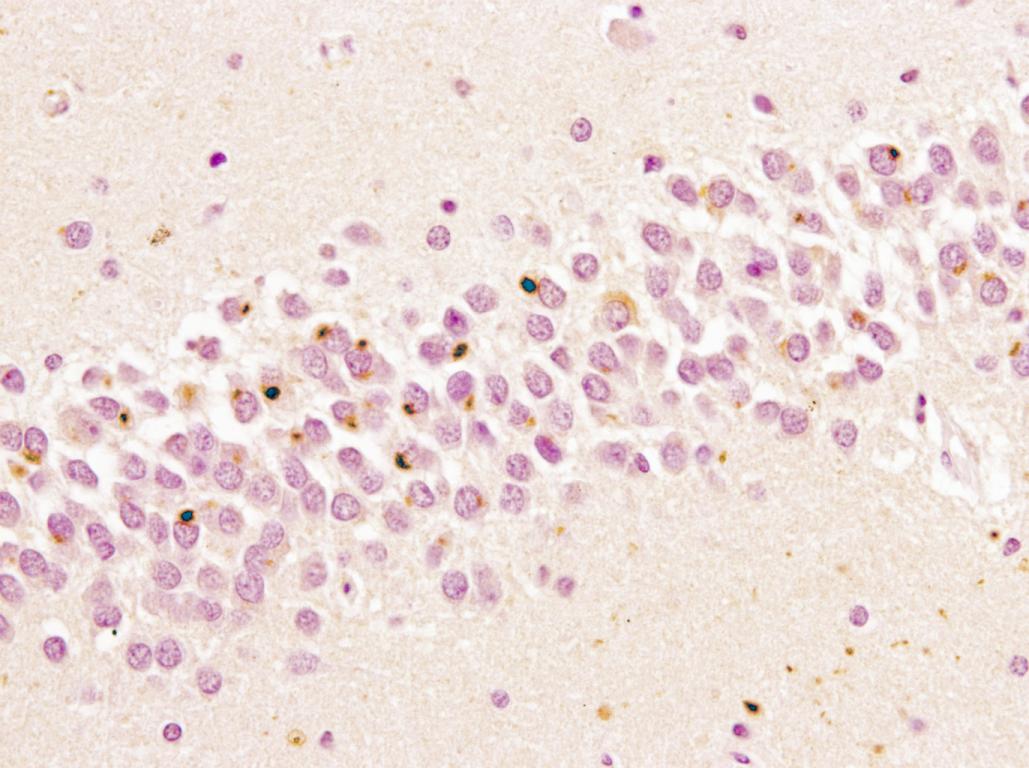
A los 4 años de inicio de los síntomas el paciente falleció haciendo donación del tejido encefálico.
Nota:
Nuestro agradecimiento a la Dra. Araceli Alonso (Hospital Ramón y Cajal. Madrid), que como responsable clínica del paciente ha contribuido a la presentación del mismo.